
SUPAC (Scale-Up and Post-Approval Changes) guidelines are regulatory frameworks developed by the U.S. Food and Drug Administration to provide guidance on changes made to pharmaceutical products after their approval. These guidelines help manufacturers manage modifications in formulation, process, equipment, site, or scale without compromising product quality, safety, or efficacy.
The primary aim of SUPAC guidelines is to ensure that any changes made during scale-up or post-approval do not adversely affect the identity, strength, quality, purity, or performance of the drug product. These guidelines also define the level of documentation, testing, and regulatory filing required for different types of changes.
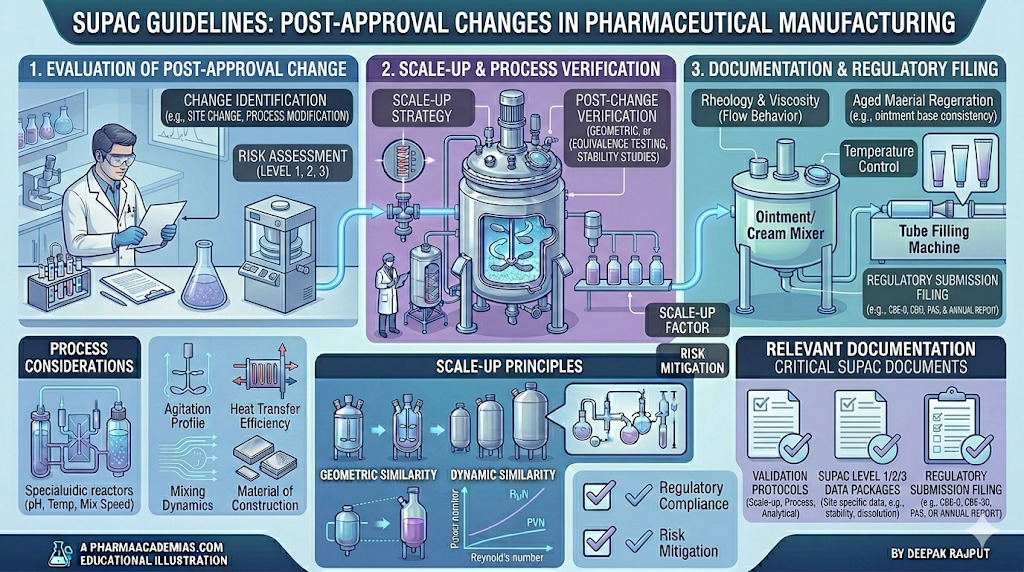
SUPAC plays a crucial role in maintaining consistency between pilot-scale, production-scale, and commercial batches.
Objectives of SUPAC Guidelines
The SUPAC guidelines are designed to standardize and regulate post-approval changes in pharmaceutical manufacturing. They aim to ensure product equivalence after any modification, reduce unnecessary regulatory burden, and facilitate continuous improvement in manufacturing processes.
They also provide a scientific basis for determining whether additional in vitro or in vivo studies are required following a change. By categorizing changes based on risk, SUPAC ensures that minor changes require minimal documentation, while major changes undergo extensive evaluation.
Categories of SUPAC Guidelines
SUPAC guidelines are classified based on dosage forms and routes of administration. The major categories include:
1. SUPAC-IR (Immediate Release Solid Oral Dosage Forms)
This guideline applies to tablets and capsules designed for immediate drug release. It addresses changes in components, composition, manufacturing process, scale-up, equipment, and site.
2. SUPAC-MR (Modified Release Solid Oral Dosage Forms)
This applies to extended-release and delayed-release formulations. Since drug release characteristics are critical, stricter evaluation and testing are required for any changes.
3. SUPAC-SS (Non-Sterile Semisolid Dosage Forms)
This guideline applies to creams, ointments, gels, and lotions. It focuses on changes in excipients, manufacturing processes, and equipment affecting product consistency and stability.
4. SUPAC-LL (Liquid Dosage Forms)
This includes oral solutions, suspensions, and emulsions. It addresses changes in formulation composition, manufacturing methods, and scale-up considerations.
Levels of Changes in SUPAC
SUPAC classifies changes into different levels depending on their potential impact on product quality.
Level 1 Changes (Minor Changes)
These are changes that are unlikely to have any significant impact on product quality or performance. Examples include minor adjustments in excipient levels or changes in equipment of the same design and operating principle.
These changes generally require minimal documentation and can often be reported in annual reports.
Level 2 Changes (Moderate Changes)
These changes may have a potential impact on product quality and therefore require additional evaluation. Examples include changes in composition beyond specified limits, changes in manufacturing process, or moderate scale-up.
Such changes typically require submission of a Changes Being Effected (CBE) supplement along with supporting data.
Level 3 Changes (Major Changes)
These changes are likely to have a significant impact on product quality, safety, or efficacy. Examples include major formulation changes, significant process modifications, or changes in manufacturing site.
These require prior approval from regulatory authorities through a Prior Approval Supplement (PAS) and extensive supporting data, including stability studies and sometimes bioequivalence studies.
Types of Changes Covered Under SUPAC
SUPAC guidelines consider several types of changes that may occur during scale-up or post-approval stages.
1. Components and Composition Changes
Changes in excipients or their quantities can affect drug release, stability, and bioavailability. SUPAC specifies acceptable limits within which changes can be made without significant impact.
2. Site Changes
These involve shifting manufacturing operations from one facility to another. The new site must demonstrate equivalent capability, equipment, and quality systems.
3. Scale-Up and Scale-Down Changes
These involve increasing or decreasing batch size. SUPAC defines acceptable scale-up limits and requires validation to ensure consistency.
4. Manufacturing Process Changes
Changes in mixing time, granulation method, drying conditions, or compression force can impact product quality and must be evaluated carefully.
5. Equipment Changes
Switching to different equipment may alter process efficiency and product characteristics. SUPAC requires demonstration of equivalence between old and new equipment.
In Vitro and In Vivo Testing Requirements
SUPAC guidelines specify testing requirements based on the level of change.
For minor changes, routine quality control tests such as dissolution, assay, and content uniformity are usually sufficient.
For moderate changes, comparative dissolution studies are required to ensure similarity between pre-change and post-change products.
For major changes, in vivo bioequivalence studies may be necessary to confirm that the modified product performs similarly to the original product.
Documentation Requirements in SUPAC
Proper documentation is essential to demonstrate that changes do not affect product quality.
Manufacturers must maintain updated Master Formula Records (MFR) and Batch Manufacturing Records (BMR) reflecting the changes. Stability data must be generated to confirm that the product remains stable under new conditions.
Process validation reports, equipment qualification documents, and analytical method validation data must also be updated. Regulatory submissions such as annual reports, CBE supplements, or PAS must include detailed justification and supporting data.
Importance of SUPAC Guidelines
SUPAC guidelines are essential for ensuring product quality and regulatory compliance during scale-up and post-approval changes. They provide a structured approach to evaluate risks associated with changes and help manufacturers implement improvements without compromising product performance.
These guidelines also facilitate faster approval of changes by providing clear regulatory pathways, thereby supporting innovation and efficiency in pharmaceutical manufacturing.
Conclusion
SUPAC guidelines are a critical component of pharmaceutical regulatory systems, ensuring that any changes made after product approval maintain consistent quality, safety, and efficacy. By categorizing changes into different levels and defining appropriate testing and documentation requirements, SUPAC provides a scientific and regulatory framework for managing scale-up and post-approval modifications effectively.
Understanding and implementing SUPAC guidelines is essential for pharmaceutical professionals involved in formulation development, manufacturing, and regulatory affairs.





