Absorption of drugs in GIT
Absorption of drugs in GIT: Absorption is one of the most fundamental and critical phases in the pharmacokinetic profile of a drug. In the context of oral drug administration, absorption refers to the movement of the drug from the gastrointestinal (GI) tract into the systemic circulation, where it can exert its therapeutic effect. The human gastrointestinal tract is a complex and dynamic system that significantly influences the absorption of orally administered medications. The efficiency of this process depends on both the physicochemical properties of the drug and the physiological characteristics of the GIT.
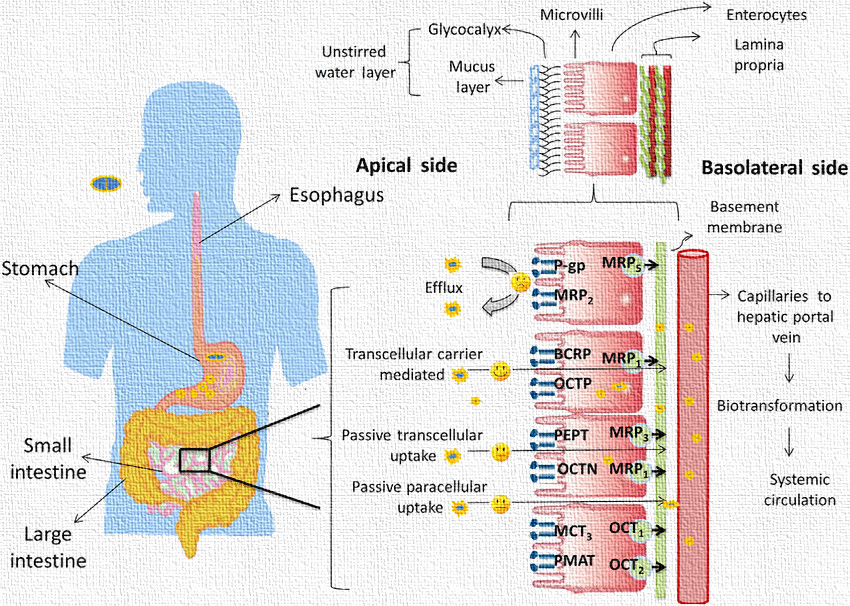
Drug absorption through the GIT is not merely a passive event; it involves a complex interplay of various mechanisms, transporters, and barriers. A clear understanding of these absorption pathways is crucial for the successful formulation of oral drug products, improvement of bioavailability, and achievement of optimal therapeutic outcomes.
Definition of Absorption
In pharmacological and biopharmaceutical terms, absorption is defined as the process by which a drug passes from its site of administration into the bloodstream. For drugs administered orally, absorption usually occurs in the small intestine, although some drugs may be absorbed in the stomach or large intestine depending on their chemical nature and formulation.
Importance of GIT in Drug Absorption
The gastrointestinal tract plays a pivotal role in the absorption of drugs due to its:
- Large surface area (especially in the small intestine due to villi and microvilli)
- Rich blood supply
- Variable pH environment (which affects drug ionization)
- Presence of transport proteins and enzymes
- Variable transit time, allowing for controlled or extended absorption
Mechanisms of Drug Absorption in GIT
The human body employs various mechanisms to facilitate the absorption of drugs across the epithelial lining of the GIT. These mechanisms are classified broadly into passive and active transport processes. The nature of the drug (such as its size, polarity, lipophilicity, and ionization state) determines which mechanism it predominantly uses.
1. Passive Diffusion (Simple Diffusion)
Passive diffusion is the most predominant and widely understood mechanism by which a large number of drugs are absorbed through the gastrointestinal tract (GIT). It refers to the movement of drug molecules across the cellular membranes of the intestinal lining without the involvement of any external energy source or transporter proteins. This process occurs simply due to a concentration gradient, where the drug moves from an area of higher concentration in the intestinal lumen to an area of lower concentration in the blood capillaries.
This mode of transport is primarily favored by drugs that are lipophilic (fat-soluble), non-ionized, and low molecular weight in nature. The cell membranes are composed of phospholipid bilayers, which allow lipophilic substances to permeate more readily than hydrophilic ones. The rate at which a drug diffuses across the membrane depends on various factors, including the surface area of the absorbing membrane, membrane thickness, concentration difference, and the partition coefficient (lipid-water solubility ratio) of the drug. These factors are mathematically represented in Fick’s Law of Diffusion.

Notably, passive diffusion is a non-saturable process, meaning it can continue as long as there is a favorable concentration gradient, regardless of how much drug is present. Also, this mechanism is non-specific, so it does not differentiate between different types of molecules. Examples of drugs absorbed via passive diffusion include aspirin, diazepam, propranolol, and theophylline. This pathway dominates because it is simple, energy-efficient, and effective for the majority of small molecule drugs.
Kye Points to remember
- Passive diffusion involves the movement of drug molecules from a region of higherconcentration (e.g., intestinal lumen) to a region of lower concentration (e.g., blood), across the epithelial membrane.
- Driving Force: Concentration gradient
- Energy Requirement: No ATP or metabolic energy required
- Selectivity: Non-selective; depends on drug’s lipophilicity and molecular size
- Favorable for: Non-ionized, lipophilic, and small molecules
- Example Drugs: Diazepam, Propranolol, Theophylline
Fick’s Law of Diffusion:

Where:
- D = Diffusion coefficient
- A = Surface area of membrane
- C1−C2 = Concentration gradient
- h = Membrane thickness
2. Facilitated Diffusion
Facilitated diffusion, also known as carrier-mediated passive transport, is a mechanism where the drug is transported across the intestinal epithelial membrane using specific carrier proteins, yet still along the concentration gradient, i.e., from a region of high concentration to low concentration. Unlike passive diffusion, which is purely driven by the drug’s physicochemical properties, facilitated diffusion requires the presence of membrane-associated proteins that recognize and bind to the drug molecules.

This mechanism is particularly important for the absorption of polar, hydrophilic, or larger molecules that cannot easily diffuse through the lipid bilayer. The carrier protein forms a reversible complex with the drug on one side of the membrane, undergoes a conformational change, and releases the drug on the opposite side. However, since it is still moving the drug along its concentration gradient, no energy (ATP) is required for the transport.
Facilitated diffusion is saturable, meaning that there is a limit to how much drug can be transported at a time because the number of carrier proteins is finite. Once all the transporters are occupied, further increases in drug concentration do not enhance the rate of absorption. Moreover, it is a selective process, as the carriers usually recognize and transport only specific types of molecules or structurally similar analogues. This makes the system susceptible to competitive inhibition by other substances using the same transporters.
Drugs and nutrients such as glucose, amino acids, thiamine (vitamin B1), and riboflavin utilize facilitated diffusion mechanisms. This type of absorption is especially significant in the small intestine where many nutrient transporters are present.
Kye Points to remember
- Involves the use of specific carrier proteins embedded in the epithelial membrane that facilitate the transport of certain drugs along the concentration gradient.
- Energy Requirement: No energy required
- Saturation: Yes; transporters can become saturated at high drug concentrations
- Specificity: Substrate-specific and susceptible to competition or inhibition
- Example Substances: Glucose, Vitamin B1 (Thiamine), certain amino acids
3. Active Transport
Active transport is a highly specialized, energy-dependent mechanism of drug absorption that enables the movement of drug molecules against their concentration gradient, i.e., from a region of lower concentration in the gut lumen to a region of higher concentration inside the enterocytes or blood capillaries. This uphill movement requires the expenditure of metabolic energy in the form of ATP (adenosine triphosphate).

Active transport mechanisms are typically mediated by specific carrier proteins, often referred to as transporters, which bind to the drug molecule and facilitate its translocation across the membrane via conformational changes. These carriers display high substrate specificity and are subject to saturation kinetics (like enzymes), meaning that the transport rate reaches a maximum when all carriers are occupied. Furthermore, active transport can be competitive, as drugs or nutrients using the same transporter can inhibit each other’s absorption.
This mechanism plays a crucial role in the absorption of drugs that mimic the structure of endogenous substances such as vitamins, amino acids, or nucleosides. For example, levodopa (L-DOPA), used in the treatment of Parkinson’s disease, is actively transported by large neutral amino acid transporters, mimicking the structure of tyrosine. Similarly, methyldopa, 5-fluorouracil, iron, and vitamin B12 are also absorbed via active transport mechanisms.
Active transport ensures that essential molecules are absorbed even when their concentrations are lower in the intestinal lumen than in the bloodstream. This feature is critical for nutrient homeostasis and therapeutic efficacy of certain drugs.
Kye Points to remember
- The transport of drug molecules against the concentration gradient (i.e., from low to high concentration), mediated by carrier proteins and utilizing ATP.
- Energy Requirement: Yes, requires ATP
- Saturation: Yes; carrier-mediated and can reach saturation
- Specificity: High substrate specificity and susceptible to drug-drug or nutrient-drug interactions
- Example Drugs: Levodopa (L-DOPA), 5-Fluorouracil, Methyldopa, Iron
4. Endocytosis and Exocytosis
Endocytosis and exocytosis are vesicular transport mechanisms that are primarily responsible for the absorption and excretion of large macromolecular substances, such as proteins, peptides, nanoparticles, and certain antibodies. These processes involve the formation of vesicles and are energy-dependent, requiring ATP.
A. Endocytosis
Endocytosis involves the engulfment of extracellular material by the cell membrane, leading to the formation of an intracellular vesicle. The membrane invaginates around the substance, encloses it, and pinches off to form a vesicle within the cell. This vesicle then may fuse with lysosomes for processing or release its contents directly into the cytoplasm. There are several types of endocytosis including pinocytosis (cell drinking), phagocytosis (cell eating), and receptor-mediated endocytosis.
This mechanism is particularly important for the absorption of very large or polar molecules that cannot cross the cell membrane by diffusion or transporters. A classic example is the absorption of vitamin B12, which requires intrinsic factor, a glycoprotein that binds to the vitamin and facilitates its endocytosis in the ileum. Similarly, nanoparticle-based drug delivery systems and biologics like insulin can exploit endocytic pathways for enhanced absorption and bioavailability.

B. Exocytosis
Exocytosis is the reverse process of endocytosis, where substances contained within intracellular vesicles are expelled from the cell into the extracellular space. The vesicles move toward the plasma membrane, fuse with it, and release their contents outside the cell. This mechanism is not primarily involved in the absorption of drugs but is significant in cellular secretion, neurotransmitter release, and immune responses.
Together, endocytosis and exocytosis represent vital cellular functions, particularly relevant to the absorption of complex or engineered therapeutic agents, including vaccines and gene therapy vectors.
5. Paracellular Transport (Pore Transport)
Paracellular transport, also known as pore transport, refers to the movement of drug molecules between adjacent epithelial cells through the tight junctions that form a seal around the cells. Unlike transcellular pathways (through cells), paracellular transport does not involve crossing the lipid bilayer but instead utilizes the aqueous pores present between cells.

This route is mainly restricted to small, hydrophilic, and low molecular weight molecules, as the tight junctions impose size and charge selectivity. The permeability of the paracellular route is generally low, especially in the small intestine, where tight junctions are narrow and tightly regulated. However, in the colon or the proximal tubules of the kidney, where tight junctions may be relatively more permissive, the contribution of this pathway can be more significant.
Unlike carrier-mediated transport mechanisms, paracellular transport is passive, driven by concentration gradients and osmotic forces, and is non-saturable and non-selective. Since it does not rely on energy or specific transporters, any suitable molecule that fits the size and solubility criteria can diffuse through this route. Drugs like mannitol, urea, and atenolol may partially utilize this mechanism for absorption.
Pharmaceutical scientists often explore ways to transiently open tight junctions using permeation enhancers or absorption promoters in order to increase paracellular transport, especially for delivering peptides and other poorly absorbed drugs.
- Pathway: Between cells, not through them
- Requirement: Drug must be small and water-soluble
- Example Substances: Urea, small ions, and water-soluble drugs like atenolol
Table of Absorption Mechanisms
| Mechanism | Energy | Carrier | Direction | Specificity | Saturation | Example Drugs |
| Passive Diffusion | No | No | High → Low | No | No | Diazepam, Aspirin |
| Facilitated Diffusion | No | Yes | High → Low | Yes | Yes | Glucose, Thiamine |
| Active Transport | Yes | Yes | Low → High | Yes | Yes | Levodopa, Methyldopa |
| Endocytosis | Yes | No | Vesicular | Yes | Yes | Vitamin B12 |
| Paracellular Transport | No | No | Between cells | No | No | Atenolol, Mannitol |
Transporters Involved in GIT Drug Absorption
Specific transporters play a major role in facilitating or limiting drug absorption:
| Transporter | Type | Role | Examples of Drugs Affected |
| P-glycoprotein (P-gp) | Efflux | Pumps drugs out of enterocytes | Digoxin, Cyclosporine |
| OATP | Influx | Mediates organic anion uptake | Statins, Fexofenadine |
| PEPT1 | Influx | Transports peptides | β-lactam antibiotics, ACE inhibitors |
| MRP | Efflux | Drug resistance protein | Methotrexate, Etoposide |
Regional Differences in GIT and Their Impact on Drug Absorption
| Region | pH Range | Characteristics | Absorption Potential |
| Stomach | 1.5–3.5 | Low surface area, acidic pH | Weak acids absorbed better |
| Duodenum | 5–6.5 | Rich blood supply, bile salts enhance solubility | High due to surface area |
| Jejunum | 6–7 | Large surface area with villi and microvilli | Most absorption occurs here |
| Ileum | 7–8 | Specialized transporters for B12 and bile salts | Moderate |
| Colon | 7–8 | Less enzymatic activity, longer retention time | Useful for sustained-release drugs |
Formulation Strategies to Enhance GIT Absorption
Pharmaceutical scientists employ several techniques to improve the absorption of poorly bioavailable drugs:
- Use of surfactants or solubilizers
- Prodrugs that become active after absorption
- Lipid-based formulations (e.g., SEDDS)
- Micronization or nanonization to improve solubility
- pH-sensitive coatings for delayed or targeted release
- Inhibitors of efflux pumps to counteract P-gp mediated drug loss
Conclusion
The absorption of drugs through the gastrointestinal tract is a multifaceted process influenced by drug properties, physiological conditions, and biochemical mechanisms. Understanding the various mechanisms of absorption, including passive diffusion, facilitated transport, active transport, endocytosis, and paracellular transport, is essential for the rational design of oral dosage forms and for predicting the bioavailability and pharmacokinetic behavior of drugs.
Biopharmaceutical studies that delve into these mechanisms provide critical insights into optimizing drug therapy, minimizing variability, and ensuring therapeutic effectiveness in the patient population.