


Introduction
Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency is one of the most extensively studied and widely distributed inherited metabolic disorders affecting humans. It is recognized as the most common enzymatic disorder of red blood cells, with an estimated global prevalence exceeding 400 million individuals. The disorder primarily affects populations living in or originating from regions where malaria has historically been endemic, including parts of Africa, the Mediterranean basin, the Middle East, South Asia, and Southeast Asia.
G6PD deficiency represents a classical example of how a genetic defect can persist in human populations due to an evolutionary survival advantage, as partial enzyme deficiency confers protection against severe Plasmodium falciparum malaria. Despite this protective effect, individuals with significant enzyme deficiency remain susceptible to potentially life-threatening hemolytic episodes when exposed to oxidative stress.

Definition
Glucose-6-Phosphate Dehydrogenase deficiency is an X-linked recessive inherited enzymatic disorder characterized by reduced or absent activity of the enzyme glucose-6-phosphate dehydrogenase in erythrocytes. This deficiency results in an impaired ability of red blood cells to counteract oxidative stress, leading to episodic or, in rare cases, chronic hemolytic anemia.
Historical Background
The clinical significance of G6PD deficiency was first recognized during the mid-20th century when hemolytic anemia was observed in certain individuals following the administration of the antimalarial drug primaquine. Subsequent biochemical investigations revealed a deficiency of G6PD enzyme activity in affected individuals, establishing a direct relationship between oxidative drugs and red blood cell destruction. Since then, G6PD deficiency has become a cornerstone topic in hematology, pharmacology, toxicology, and pharmacovigilance.
Epidemiology
The prevalence of G6PD deficiency varies widely across different populations and ethnic groups. It is particularly common in:
- Sub-Saharan Africa
- Mediterranean regions such as Italy and Greece
- Middle Eastern countries
- The Indian subcontinent
- Southeast Asia
The condition predominantly affects males, as they possess only one X chromosome, whereas females may be asymptomatic carriers or partially affected due to random X-chromosome inactivation (lyonization). In certain populations, up to 10–25% of males may carry G6PD variants.
Genetics and Inheritance Pattern
G6PD deficiency follows an X-linked recessive pattern of inheritance. The gene encoding the G6PD enzyme is located on the long arm of the X chromosome at position Xq28.
- Males (XY) with a defective gene are fully affected.
- Females (XX) may be carriers, partially deficient, or rarely fully affected, depending on the pattern of X-chromosome inactivation.
More than 400 genetic variants of the G6PD gene have been identified, resulting in varying degrees of enzyme deficiency and clinical severity.
WHO Classification of G6PD Variants
The World Health Organization has classified G6PD variants into five classes based on enzyme activity and clinical presentation:
- Class I: Severe deficiency with chronic nonspherocytic hemolytic anemia
- Class II: Severe deficiency with intermittent hemolysis
- Class III: Moderate deficiency with hemolysis under stress
- Class IV: Normal enzyme activity
- Class V: Increased enzyme activity (rare, clinically insignificant)
Biochemical and Physiological Role of G6PD
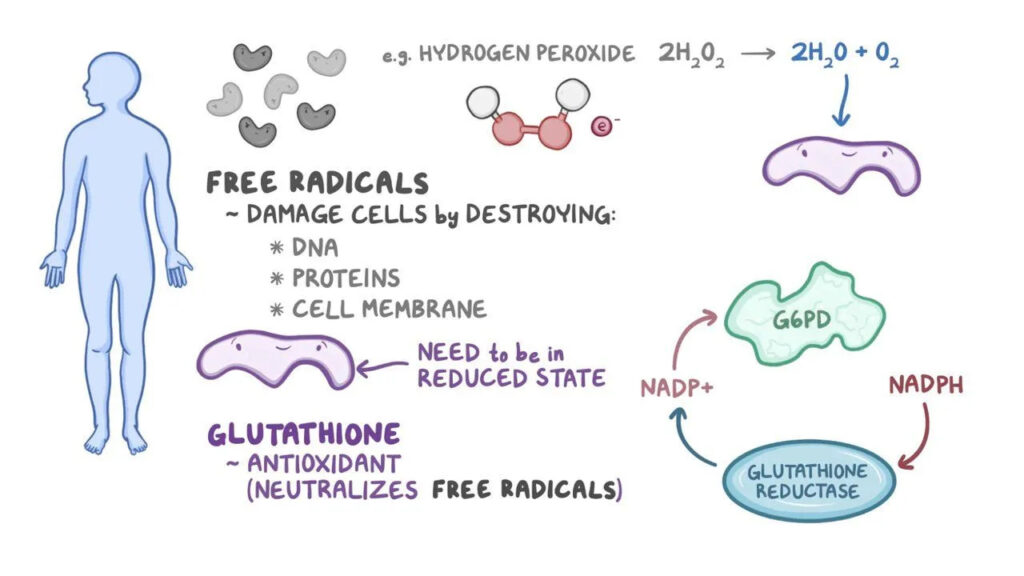
Glucose-6-phosphate dehydrogenase is the first and rate-limiting enzyme of the pentose phosphate pathway, also known as the hexose monophosphate shunt. This metabolic pathway is essential for the generation of nicotinamide adenine dinucleotide phosphate in its reduced form (NADPH).
In erythrocytes, which lack mitochondria, the pentose phosphate pathway is the only source of NADPH. NADPH plays a crucial role in maintaining glutathione in its reduced form (GSH), which protects hemoglobin and red cell membranes from oxidative damage caused by reactive oxygen species.
Pathophysiology
In individuals with G6PD deficiency, reduced enzyme activity leads to diminished NADPH production. As a result, glutathione cannot be adequately regenerated in its reduced form. This biochemical imbalance makes red blood cells extremely vulnerable to oxidative injury.
Oxidative stress causes denaturation of hemoglobin, leading to the formation of Heinz bodies, which attach to the red cell membrane. These damaged cells are either destroyed within the circulation (intravascular hemolysis) or removed by macrophages in the spleen (extravascular hemolysis), resulting in hemolytic anemia.
Triggers of Hemolytic Episodes
Hemolysis in G6PD deficiency is typically episodic and occurs following exposure to oxidative stressors.
Common Triggers Include:
Drugs
- Antimalarials such as primaquine
- Sulfonamide antibiotics
- Nitrofurantoin
- Dapsone
- Certain quinolones
- High-dose aspirin
- Methylene blue
Foods
- Fava beans, which contain oxidant compounds (favism)
Infections
- Bacterial and viral infections that increase endogenous oxidative stress
Chemicals
- Naphthalene found in mothballs
Clinical Manifestations
Neonatal Period
G6PD deficiency is an important cause of neonatal hyperbilirubinemia, which may progress to kernicterus if left untreated.
Children and Adults
Clinical features during acute hemolytic episodes include:
- Sudden onset of anemia
- Fatigue and generalized weakness
- Pallor and jaundice
- Dark or cola-colored urine
- Abdominal or back pain
- Shortness of breath and tachycardia
Chronic Hemolysis
Rarely, individuals with severe variants may experience chronic hemolytic anemia, leading to splenomegaly and pigment gallstone formation.
Laboratory Diagnosis
Laboratory evaluation reveals:
- Decreased hemoglobin levels
- Elevated reticulocyte count
- Increased indirect bilirubin
- Raised lactate dehydrogenase (LDH)
- Reduced haptoglobin levels
Peripheral blood smear may show:
- Heinz bodies (on supravital staining)
- Bite cells and blister cells
The definitive diagnosis is established by quantitative G6PD enzyme activity assays.
Management and Treatment
There is no curative treatment for G6PD deficiency. Management focuses on prevention and supportive care.
- Avoidance of known triggers
- Prompt treatment of infections
- Blood transfusions in severe hemolysis
- Phototherapy or exchange transfusion in neonates
Patient education and genetic counseling play a central role in long-term management.
Pharmacological and Clinical Importance
G6PD deficiency has significant implications in pharmacotherapy and pharmacovigilance, as many commonly used drugs can precipitate hemolysis. Healthcare professionals must screen patients and prescribe medications cautiously.
Prognosis
With proper awareness and avoidance of triggers, individuals with G6PD deficiency generally have a normal life expectancy and good quality of life.
Conclusion
Glucose-6-Phosphate Dehydrogenase deficiency is a genetically inherited enzymatic disorder that highlights the delicate balance between human genetics, environmental exposure, and drug therapy. Although often asymptomatic, exposure to oxidative stress can result in severe hemolytic anemia. Early diagnosis, rational drug use, and patient education remain the cornerstones of effective management.